At last, one of the papers investigating H5N1 influenza transmission in ferrets has been published in the journal Nature yesterday. To recap the controversy briefly: news of experimental studies investigating transmissibility of avian H5N1 influenza hit the news this fall, igniting a fierce debate about biosecurity, “dual use” research, and the damage that censorship can have on scientific advancement. An advisory group, the NSABB, recommended partial censorship of the data, perhaps believing that redacting specific data from the publications would prevent information on how to generate a highly pathogenic mammalian transmissible virus from getting into the hands of bioterrorists or others incapable of handling such viruses safely. However, after some new data or clarification of data presented in a revised version of the submitted manuscript, the NSABB recommended publication in full.
Avian H5N1 influenza virus has caused sporadic infections in humans who have close contact with infected animals. Human-to-human transmission has not been observed. But could an H5 virus mutate or reassort, allowing human-to-human transmission?
One thing that has been lost in this whole controversy is that this study is actually a great demonstration and application of evolution. How does a virus switch to a new host and transmit efficiently between individuals? Start with a diverse population that varies in a particular trait (in this case, ability to bind the human receptor). Put it through selective pressure. This occurred in several steps: first, select for viruses that can bind the human receptor in vitro. From those that bind, select ones that can efficiently replicate in the respiratory tract of the animal. Finally, take the efficient replicators and allow for transmission. At each step of selection, mutations that naturally occur during viral replication further diversify the population resulting in variants that possess the desired property. Those variants get selected for the next experiment.
This study focused on one particular influenza virus protein, hemagglutinin (HA). HA is on the surface of the virion and is what the virus uses to attach to the host cell, the first step in viral infection. HA of human influenza viruses bind a sugar on the surface of human cells, which is slightly different from that found in the avian respiratory tract. Avian viruses, of course, bind to the form found in birds. H5 shows a strong preference for binding the avian receptor, so Karaoka et al were interested in finding out if H5 could change to recognize the human receptor, allowing more efficient transmission between mammals.
To address this, they began with a mutagenesis technique to introduce random mutations in the globular head (the receptor binding part) of HA, then used an in vitro approach to select for mutants that bound the human receptor (selection step 1). Through this process they identified three H5 variants that gained the ability to bind the human receptor while maintaining the ability to bind the avian receptor, and one that switched specificity completely to the human receptor. Several of the mutations identified in this study had already been shown in previous studies to be important in receptor specificity.
To test if the variant H5s conferred binding to human receptors in vivo, sections of human tracheal tissue were exposed to the viruses and only two were able to infect (selection step 2). This suggests the virus can infect human epithelium of the upper respiratory tract.
Next, the two remaining variants were used to infect ferrets. Both replicated in ferret respiratory tracts, but one replicated to higher levels. When they sequenced the virus that they isolated from that ferret, it was different from what they had put in: a new mutation had appeared. This new mutation presumably confers the property of better replication in the ferret respiratory tract, so it outgrew the original input virus (selection step 3).
Using this new virus (now with a total of 3 mutations in H5), transmission was tested in ferrets. Compared to the original H5, which did not transmit via aerosol, the 3-mutation variant did transmit, although between only 2 of 6 animal pairs. Again, they sequenced the virus that was present in the contact animals and found that it was different than what had gone in to the inoculated animals. Yet another mutation had appeared (selection step 4). This additional mutation appears to enhance transmission: the new virus, now with 4 mutations, transmitted more quickly and between more pairs of animals than the 3-mutation virus. Although the virus can transmit, none of the infected animals died, but they did show pathology at the site of infection.
So what does this all mean? The best model available for influenza transmission studies is ferrets. Ferrets aren’t humans, so its important to keep in mind that this is a model that helps us understand what viral or host factors are involved in aerosol transmission in these mammals, and maybe, but not necessarily, in humans. Since we don’t know what is necessary for human-to-human transmission, it is valuable to have an animal model to give us some ideas of what to look for. It can provide some good hypotheses on what mediates transmission in humans, which would then have to be further tested. Obviously, specificity for the human receptor is necessary, but the mutations identified tell us more than that. Mutations that change specificity are not sufficient for transmission. It turns out that those mutations also decrease the stability of the HA protein. The additional mutations acquired through the selection steps compensate for that, and enhance stability. So now we know that HA stability is important in influenza transmission. Between ferrets. That’s probably true for humans too, but it would need to be tested.
If you are still reading, you are obviously procrastinating, and are probably avoiding studying for your final exam. But here are some more thoughts on the controversy overall. This is a really interesting paper, with nothing particularly frightening or worrisome about it. Certainly not any more so than other papers doing similar work that were published without so much controversy. If “dual use” research needs to be regulated, it needs to be done before the work is done, not after. If the NSABB was concerned about this kind of research, why only express concern once the experiment succeeds? In my intro biology class, we read another paper, published in 2005, which was addressing the exact same question, in an almost identical way. The difference is that they failed to make a transmissible virus. If there is a concern about this kind of research, a concern that it is too risky to do these kinds of experiments, shouldn’t the alarm have been raised regardless of the outcome? It just doesn’t make sense to me why it suddenly became so concerning. If anything good has come of this controversy, it is the widespread discussion that this has stimulated on the importance of open communication of scientific data, the importance of not censoring in science. Ironically, had we all been given access to the data, like through a journal publication, it would have been apparent that there wasn’t anything to be concerned about.

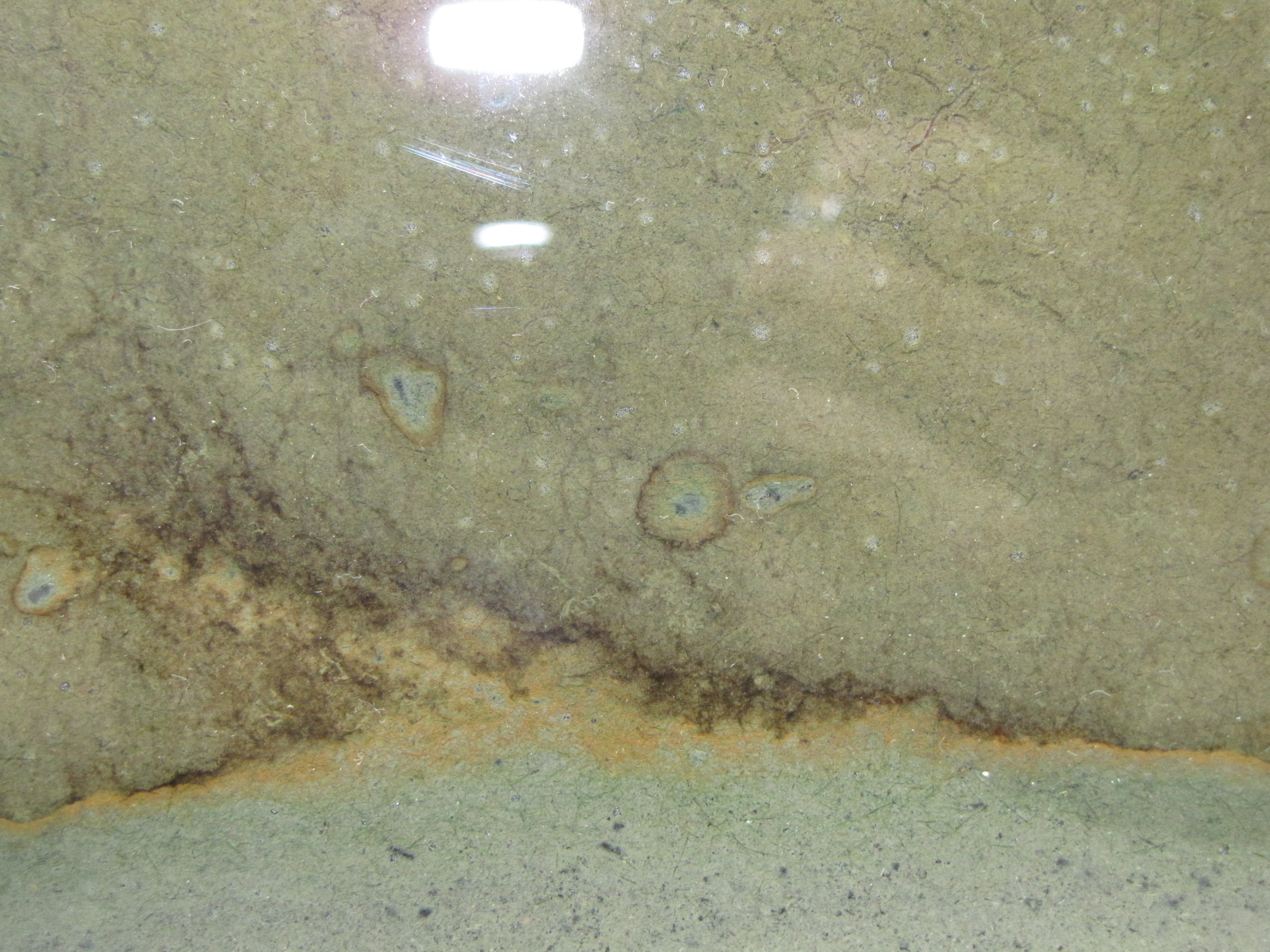
 The level of the mud dropped overnight. This could be due to settling of the mud and/or release of gas bubbles. There are some water-filled gaps, and the water level is lower. There is now air above the entire length of the water surface, instead of just at the opening of the panel (top left). Note that there is also a left to right gradient in the water, which was already present before settling. Near the opening, there is brownish/red colonies growing on the walls, probably aerobes (requires oxygen) explaining why its not present further from the opening. The water is murky from left to right, perhaps microbes that can tolerate low levels of oxygen, up to a point near the right side where the water layer is still clear.
The level of the mud dropped overnight. This could be due to settling of the mud and/or release of gas bubbles. There are some water-filled gaps, and the water level is lower. There is now air above the entire length of the water surface, instead of just at the opening of the panel (top left). Note that there is also a left to right gradient in the water, which was already present before settling. Near the opening, there is brownish/red colonies growing on the walls, probably aerobes (requires oxygen) explaining why its not present further from the opening. The water is murky from left to right, perhaps microbes that can tolerate low levels of oxygen, up to a point near the right side where the water layer is still clear.