Now that we’ve analyzed both the  H-NMR and the
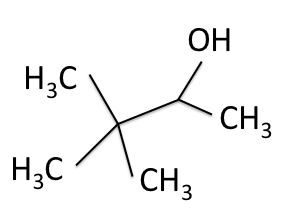
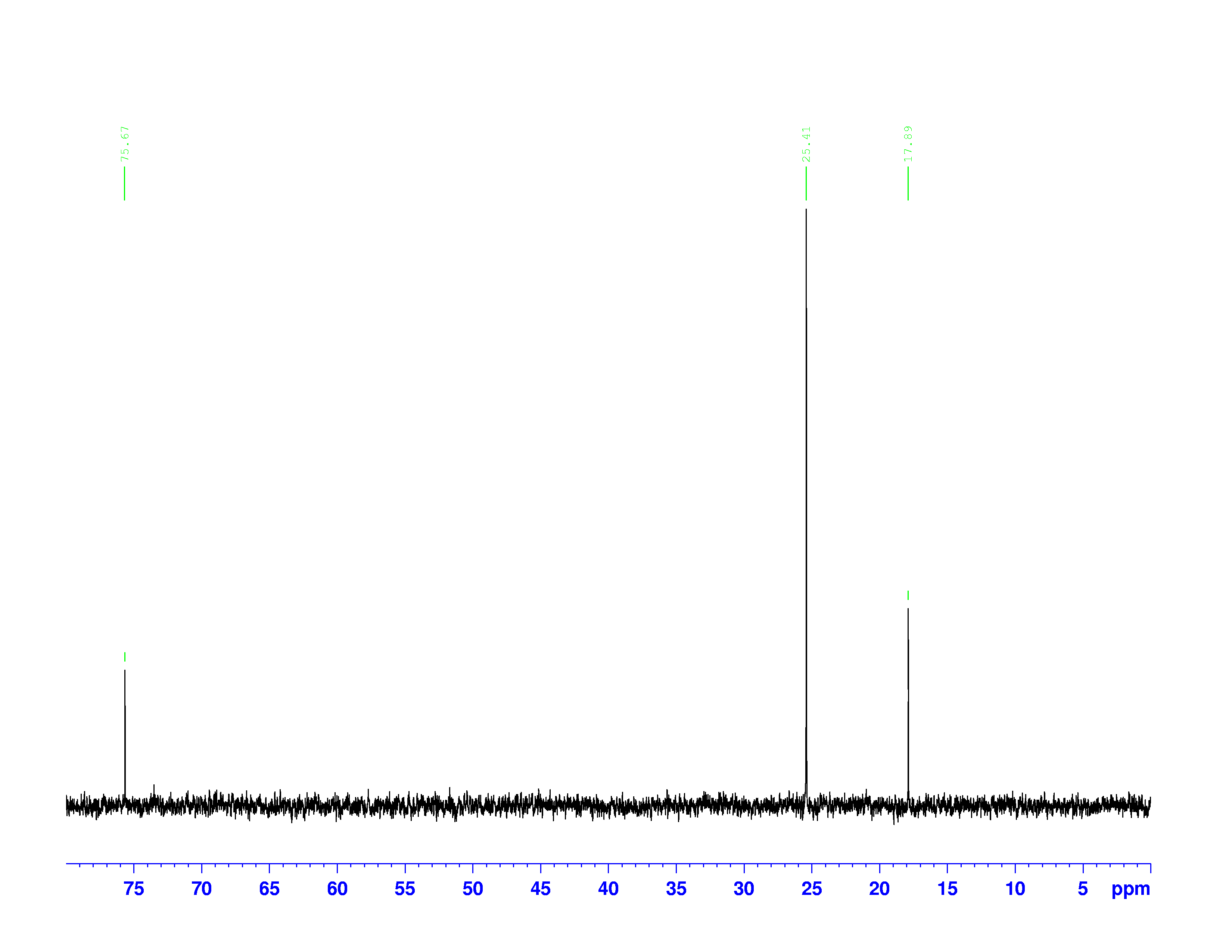
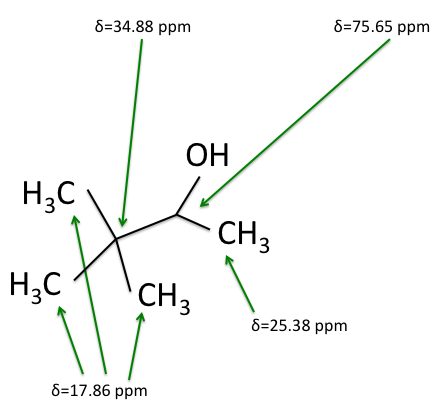
H-NMR and the  C-NMR spectra, we can see how the results come together and give the structure of 3,3-dimethyl-2-butanol. Below we bring back the two figures that show the labeled hydrogens and carbons of the molecule:
C-NMR spectra, we can see how the results come together and give the structure of 3,3-dimethyl-2-butanol. Below we bring back the two figures that show the labeled hydrogens and carbons of the molecule:


If we did not already know the identity of the molecule, we could have determined it using only the four spectra previously shown–the H-NMR, the C-NMR, the DEPT-90, and the DEPT-135–by following the guidelines laid out in the previous posts. Peaks on the four spectra work together to give bits of information about pieces of the molecule, and those pieces are put back together at the end of the analysis.
To summarize, we began by taking advantage of the magnetic properties of H and C nuclei to learn about the environment of each nucleus in the molecule. Radio waves were pulsed at the sample to record small changes in the resonant Larmor frequency of the nuclei. The change happened because electrons around each nucleus became a current when influenced by the NMR’s external field, and generated their own fields in the opposite direction according to Lenz’s Law. This new, effective magnetic field resulted in changes in the Larmor frequency of each nucleus. It is these changes that are displayed on the spectra as chemical shifts. The chemical shift is measured in ppm of the original Larmor frequency of the nucleus.
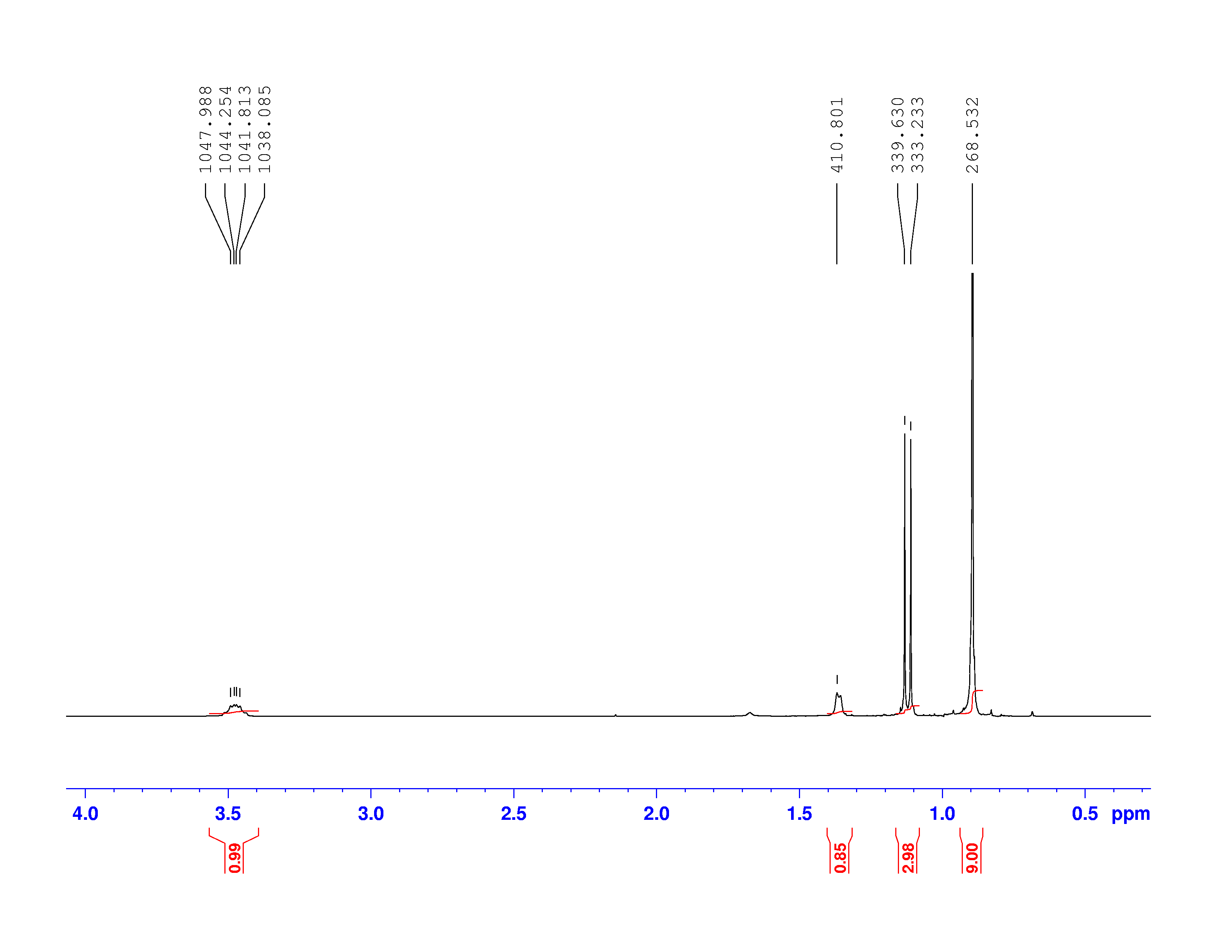
Using the Larmor frequency equation below, and the shifted frequencies, we calculated the shielding factor  for each nucleus. The shielding factor is another measure of the relative electron density around each nucleus.
for each nucleus. The shielding factor is another measure of the relative electron density around each nucleus.
(1) 
Combining the information from the spectra gives the structure of 3,3-dimethyl-2-butanol, which is shown again below.
 is 300 MHz.
is 300 MHz.  magnetic field. According to Jacobsen in “NMR Spectroscopy Explained,” the Larmor frequency of unaltered
magnetic field. According to Jacobsen in “NMR Spectroscopy Explained,” the Larmor frequency of unaltered  , is 672.650
, is 672.650 We bring back the equation for Larmor Frequency:
We bring back the equation for Larmor Frequency: C nuclei. It comes down to the composition of the nucleus. Only nuclei with nonzero spin are magnetically active. Spin is a type of angular momentum intrinsic to subatomic particles. Particles with nonzero spin can have magnetic moments that can be influenced by a magnetic field. Any nucleus with an even number of protons and an even number of neutrons will not be magnetically active because its spin is 0 and it has no magnetic moment. Carbon-12 has 6 protons and 6 neutrons, so it can’t be studied with NMR. Carbon-13 has 6 protons and 7 neutrons, so we can study it with NMR. The structure and
C nuclei. It comes down to the composition of the nucleus. Only nuclei with nonzero spin are magnetically active. Spin is a type of angular momentum intrinsic to subatomic particles. Particles with nonzero spin can have magnetic moments that can be influenced by a magnetic field. Any nucleus with an even number of protons and an even number of neutrons will not be magnetically active because its spin is 0 and it has no magnetic moment. Carbon-12 has 6 protons and 6 neutrons, so it can’t be studied with NMR. Carbon-13 has 6 protons and 7 neutrons, so we can study it with NMR. The structure and 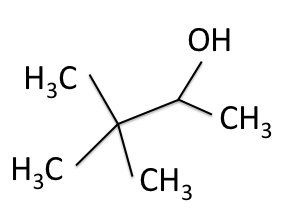
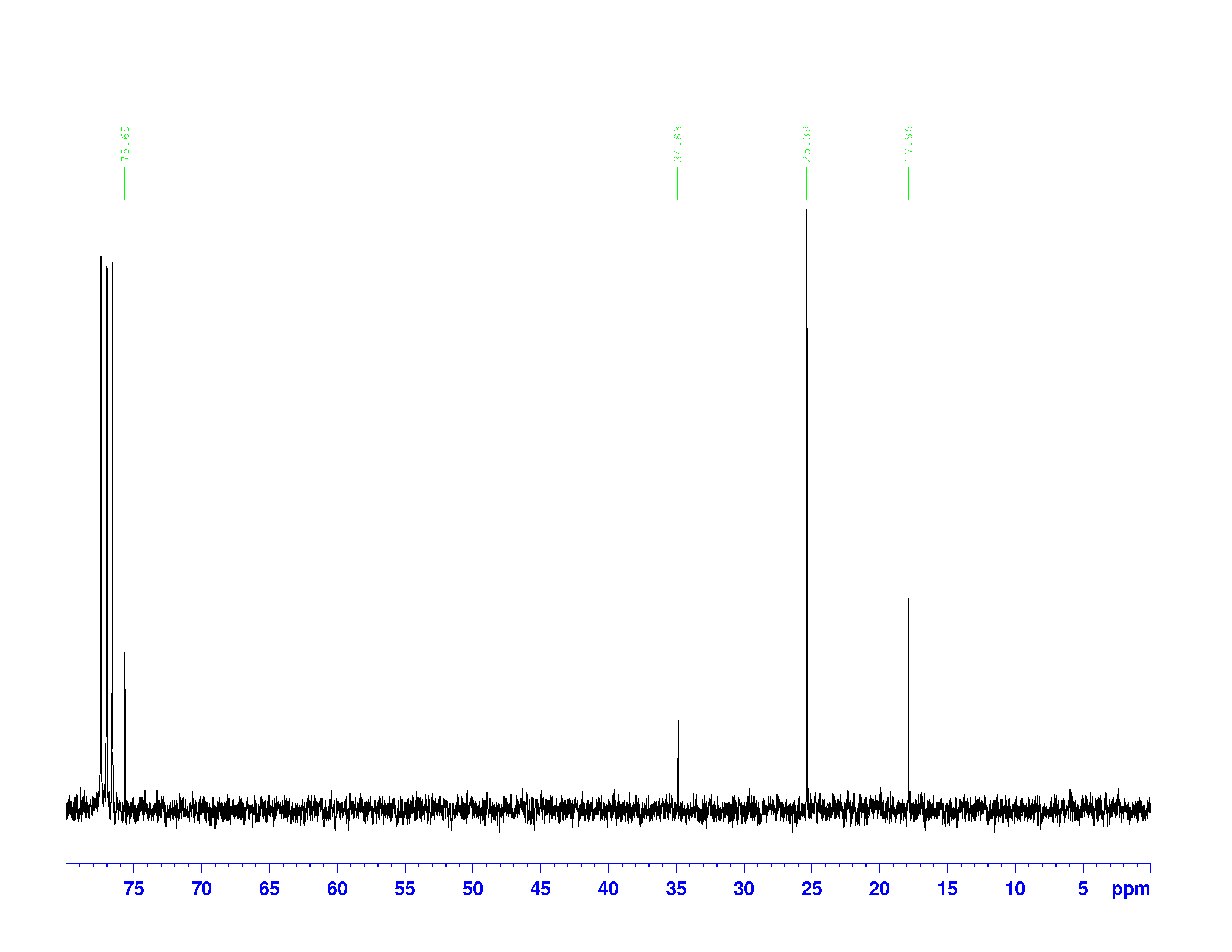
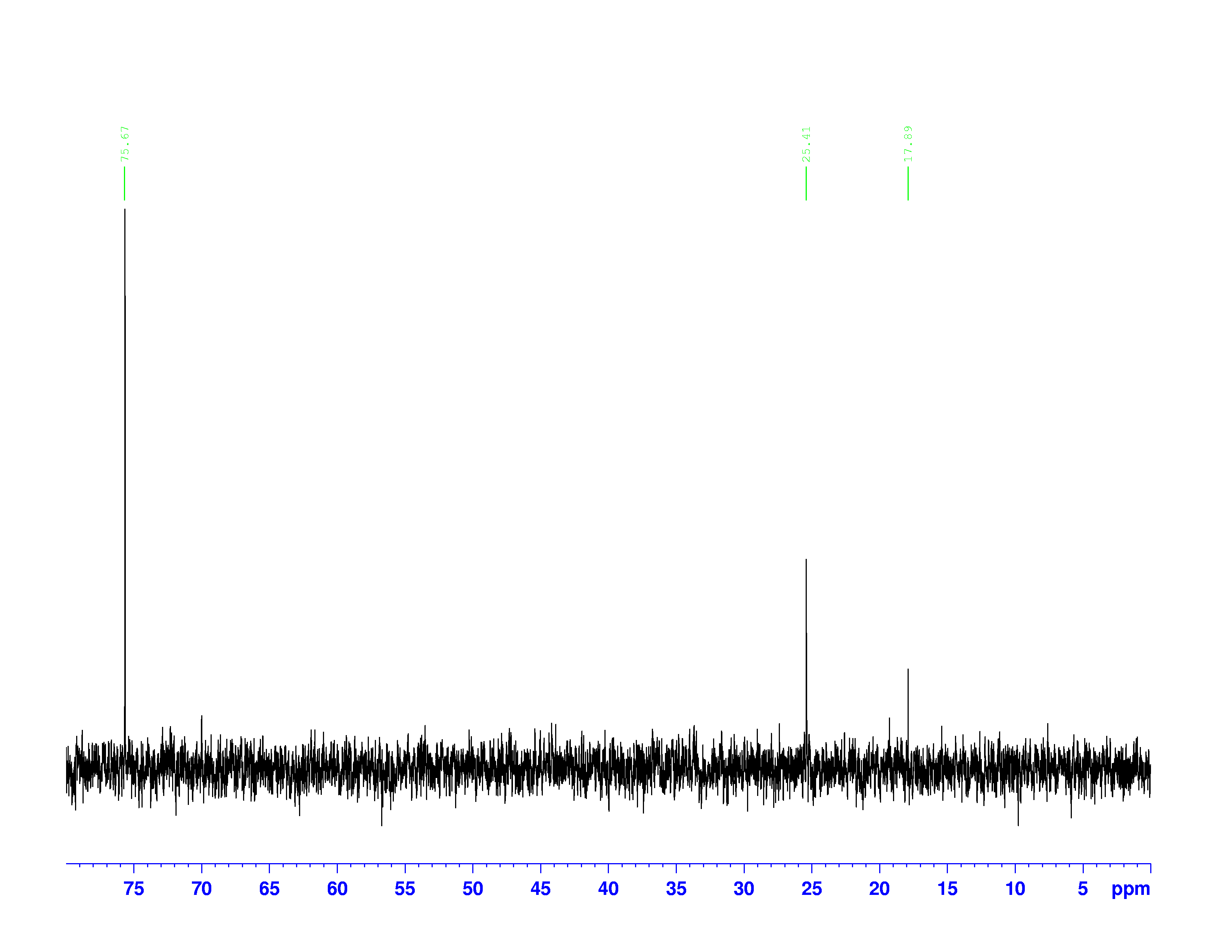
 , used to dissolve the sample. Excluding that triplet, the molecule has four different kinds of carbon. To help us find what carbons are represented here, we turn to the other two spectra of interest. They are called the DEPT-90, and the DEPT 135. DEPT stands for Distortionless Enhancement by Polarization Transfer. The technique hits the sample with five successive radio pulses designed to excite either hydrogen or
, used to dissolve the sample. Excluding that triplet, the molecule has four different kinds of carbon. To help us find what carbons are represented here, we turn to the other two spectra of interest. They are called the DEPT-90, and the DEPT 135. DEPT stands for Distortionless Enhancement by Polarization Transfer. The technique hits the sample with five successive radio pulses designed to excite either hydrogen or 

 ) doped with magnesium oxide (MgO). The magnesium oxide is there to prevent optical damage that can occur with high energy light in the bluish green frequencies. The Index of refraction of this crystal is
) doped with magnesium oxide (MgO). The magnesium oxide is there to prevent optical damage that can occur with high energy light in the bluish green frequencies. The Index of refraction of this crystal is  = 2.29 and the linear electro optic coefficient r = 30.9. I could not find any information on R the quadratic electro optic coefficient. Inside the EoM, the crystal has a parallel plate capacitor across it as shown below.
= 2.29 and the linear electro optic coefficient r = 30.9. I could not find any information on R the quadratic electro optic coefficient. Inside the EoM, the crystal has a parallel plate capacitor across it as shown below.


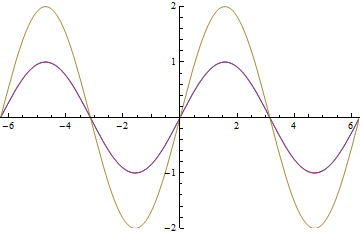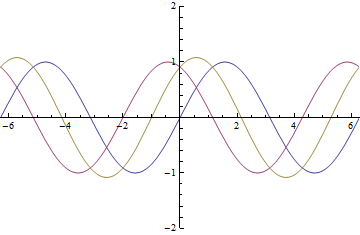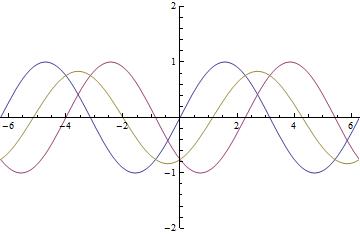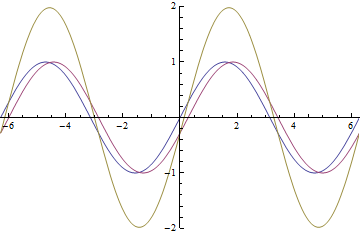
 as it passes through the EoM. The yellow wave is resultant amplitude modulation.
as it passes through the EoM. The yellow wave is resultant amplitude modulation.
 . A quick calculation using:
. A quick calculation using:



 , where
, where  is the number of peaks. The definition of “nearby” is usually 1 carbon atom over in the molecule from the one the original hydrogen is attached to.
is the number of peaks. The definition of “nearby” is usually 1 carbon atom over in the molecule from the one the original hydrogen is attached to.





{kind=link}